
Ein langer Prozess: So läuft die Zulassung neuer Medikamente ab
Vor dem Zulassungsantrag
Bevor es mit der Entwicklung des eigentlichen Medikaments losgehen kann, gibt es in der Regel eine lange Forschungsperiode. Denn nur sehr selten wird ein Wirkstoff "per Zufall" entdeckt. Üblicherweise arbeiten Wissenschaftler gezielt daran, Medikamente für bestimmte Krankheiten zu finden oder Therapien zu verbessern. Diese Forschungsperiode beinhaltet daher zunächst viele Anpassungen einzelner Substanzen, die als Wirkstoff für ein neues oder verbessertes Medikament infrage kommen.[2]
Von 5.000 bis 10.000 anfänglich untersuchten Substanzen kommen im Durchschnitt nur neun in ersten Studien mit Menschen zur Erprobung, und nur eine erreicht tatsächlich später den Markt.[2]
Nach der vorklinischen Phase kann der Wirkstoff in sogenannten klinischen Studien erstmals an Menschen angewendet werden. Das ist der letzte Schritt vor der Zulassung eines Medikaments.[3]
Erst wenn alle Studien und Tests erfolgreich waren, die Wirksamkeit und Verträglichkeit des Medikaments für die jeweilige Patientengruppe also ausreichend überprüft sind, kann die Zulassung beantragt werden. Von 5.000 bis 10.000 untersuchten Substanzen wird durchschnittlich für etwa eine Substanz ein Zulassungsantrag gestellt.[2]
Der Zulassungsantrag: Prozess in der EU
Der Prozess für den Zulassungsantrag variiert je nach Land, in dem das Medikament zugelassen werden soll. Um ein Medikament in Europa verfügbar zu machen, gibt es unterschiedliche Wege: Der Antrag kann zum einen bei einer nationalen Behörde gestellt werden. In Deutschland sind dies das Bundesinstitut für Arzneimittel und Medizinprodukte, kurz BfArM, in Bonn und das Paul-Ehrlich-Institut, kurz PEI, in Langen bei Frankfurt am Main. Welche der beiden deutschen Behörden zuständig ist, wird im Arzneimittelgesetz geregelt. So ist das PEI zum Beispiel zuständig für Allergene, Impfstoffe und gentechnisch hergestellte Blutbestandteile.[4] Ein Antrag bei einer nationalen Behörde bedeutet, dass das Medikament anschließend nur in dem jeweiligen Land vermarktet werden kann.[5] Wenn es um die Zulassung für die gesamte EU geht, wird der Antrag jedoch meist direkt bei der Europäischen Arzneimittelagentur, kurz EMA, in Amsterdam gestellt. Bei innovativen Arzneimitteln und Therapien, auch bei Orphan Drugs, ist dieses sogenannte „zentralisierte Verfahren“ sogar verpflichtend.[5]
Die EMA hat die Aufgabe, Human- und Tierarzneimittel zu beurteilen und zu überwachen und so die Gesundheit von Menschen und Tieren zu schützen. Außerdem fördert sie Innovation und Forschung in der pharmazeutischen Industrie, zum Beispiel mit der wissenschaftlichen Beratung bei der Durchführung klinischer Studien.[6]
Beim Zulassungsverfahren über die EMA hat der Ausschuss für Humanarzneimittel, kurz CHMP (aus dem Englischen: Committee for Human Medicinal Products), eine besondere Rolle: Seine Aufgabe ist es, die Antragsunterlagen wissenschaftlich zu bewerten. Dabei prüft das CHMP alle vom Pharmaunternehmen zum neuen Medikament zusammengestellten Studiendaten und Informationen. Oftmals sind das mehr als 500.000 Seiten. Geprüft werden unter anderem die Qualität und Wirksamkeit des Medikaments, aber auch die Unbedenklichkeit und Umweltverträglichkeit.[7] Im CHMP finden sich Wissenschaftler aus allen Ländern der EU, genauer gesagt aus den jeweiligen Zulassungsbehörden. Für Deutschland sind das zum Beispiel Mitarbeiter des BfArM und des PEI.[7]
Die Bearbeitung des Antrags bis zur endgültigen Zulassung des Medikaments durch die Europäische Kommission dauert im Schnitt 13 Monate.[2] In dieser Zeit kann das CHMP auch Rückfragen an das jeweilige Pharmaunternehmen stellen. Der Antragsteller hat dann in der Regel drei Monate Zeit, um diese Rückfragen zu beantworten.
Wenn alle Fragen geklärt wurden, erstellt das CHMP ein Gutachten, das eine positive oder negative Empfehlung zur Zulassung enthält. Diese Empfehlung ist die Basis für die eigentliche Zulassung und damit von sehr großer Bedeutung.
Bei positiver Empfehlung kommt es in der Regel zur Zulassung des neuen Medikaments durch die europäische Kommission – für den gesamten europäischen Raum im Normalfall für zunächst fünf Jahre. [8] Nach Ablauf dieser Zeit kann die Zulassung nach erneuter Prüfung verlängert werden.[6] In Deutschland kann der Hersteller das Präparat unverzüglich in den Markt bringen. In vielen anderen Ländern Europas ist das erst nach Verhandlungen mit Gesundheitsbehörden oder Kassenvertretern über die Kostenerstattung möglich.[2] Zudem muss in allen Ländern vorab der sogenannte „Linguistic Review“, eine sichere und gründliche Übersetzung der Fachinformation und Gebrauchsinformation, erfolgen.
Vom Projektbeginn, also der Forschungsperiode, bis zur Markteinführung eines Medikaments vergehen wie bereits erwähnt in der Regel mehr als 13 Jahre. Für die Unternehmen bedeutet das sehr hohe Kosten von bis zu 1,6 Milliarden US-Dollar, in die auch die fehlgeschlagenen Prozesse mit eingerechnet sind.[2] Umso wichtiger ist es für sie, dass den Patienten am Ende des Prozesses ein wirksames und sicheres Medikament zur Verfügung steht – in der Hoffnung, dass es dann möglichst vielen Patienten helfen kann.
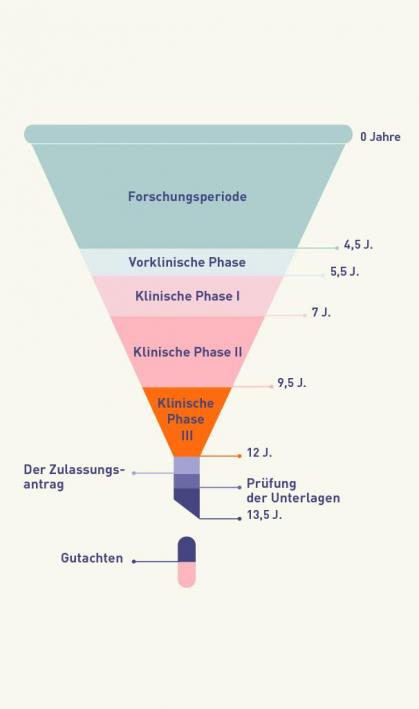
So lange dauern die Phasen des Zulassungsprozesses neuer Medikamente im Durchschnitt in Jahren.
Der Zulassungsprozess im Überblick
- Forschungsperiode: Wissenschaftler suchen gezielt nach einem Wirkstoff zur Behandlung oder Heilung einer Krankheit. Die Phase geht mit vielen Änderungen an den einzelnen möglichen Substanzen einher.
- Vorklinische Phase: Die möglichen Wirkstoffkandidaten werden auf ihre Wirkung und Verträglichkeit geprüft.
- Klinische Phase I: Der Wirkstoff wird erstmals an wenigen gesunden Menschen getestet. Die Forscher prüfen hier, was die Substanz mit dem Körper macht, beispielsweise ob und welche Nebenwirkungen auftreten.
- Klinische Phase II: Es finden Untersuchungen der Wirkung und Verträglichkeit des Medikaments mit wenigen kranken Menschen über einen kurzen Zeitraum statt. Hier wird zudem getestet, wie hoch die optimale Dosierung des Medikaments ist.
- Klinische Phase III: Es folgt die Erprobung des Medikaments an vielen kranken Menschen für einen längeren Zeitraum. In dieser Phase können auch seltenere Nebenwirkungen erkannt werden.
- Der Zulassungsantrag: Erst wenn alle Studien und Tests erfolgreich waren, kann die Zulassung beantragt werden. Der Prozess für den Zulassungsantrag variiert je nach Land und wird in Deutschland beim Bundesinstitut für Arzneimittel und Medizinprodukte (BfArM) oder dem Paul-Ehrlich-Institut (PEI) gestellt. Wenn es um die Zulassung in der gesamten EU geht, ist hingegen die Europäische Arzneimittel-Agentur (EMA) zuständig.
- Prüfung der Unterlagen: In der EU prüft der Ausschuss für Humanarzneimittel (CHMP) der EMA die Antragsunterlagen, welche vom Pharmaunternehmen eingereicht wurden. Geprüft werden die Qualität, Wirksamkeit, Unbedenklichkeit und Umweltverträglichkeit des Medikaments.
- Gutachten: Wenn alle Unterlagen geprüft und alle Fragen geklärt wurden, erstellt das CHMP ein Gutachten mit einer positiven oder negativen Empfehlung zur Zulassung. Dies ist die Basis für die eigentliche Zulassung durch die Europäische Kommission.
DE-20-2200202


